Once a medicinal product receives the Marketing Authorization approval and the product is ready to be commercialized, further regulatory requirements need to be taken into account throughout the product life-cycle to ensure a compliant, sustained, and successful product use.
Up to 70% of the post-approval activities of a medicinal product, including pharmacovigilance, could involve maintenance. Thus, product maintenance is a field with a large potential for improvement in both management and execution.
Asphalion has many years of experience in product Life-Cycle Management and is responsible for the portfolio maintenance of large, medium-sized and small pharmaceutical and biotech companies.
As a team of specialists in current medicinal regulations in EU, USA and many other countries, Asphalion can assist you in all medicinal product life-cycle management needs.
Life-Cycle Management Solutions
Regulatory strategy and maintenance support throughout product life-cycle post-approval:
Expert support on responses to Deficiency Letters.
Dossier consolidation of legacy dossiers (Mod. 2.3 and 3) for eCTD publishing.
Ad hoc regulatory strategy.
Pharmacovigilance strategy and operations that responds to legal requirements and best enhances the safety profile of your product in real world application:
Management and follow-up of data and evidence generation post-approval.
Ensuring complete and constant subjection to compliance based on specific regional regulations:
Readability tests for further EU registration processes.
Data retrieval for price and reimbursement strategies.
Price and reimbursement for new medicinal products and for generic products in Spain.
Marketing Authorization transfer management.
Support in UK with new MHRA requirements:
Medical device related post-commercialization regulation:
Validation and qualification of medical devices manufacturing process, equipment and facilities.
SaMD validation for medical devices: Standalone software as medical device (SaMD). Computerized Systems as part of a medical device.
Complete support in Health Technology Assessment (HTA).
Schedule a free meeting!
Discuss your case with our experts and receive a valuable feedback
Share
Why Asphalion?
At Asphalion, we have many years of experience in product Life-Cycle Management and are responsible for product maintenance of large, medium-sized and small pharmaceutical and biotech companies. We provide this maintenance activities for multiple regional contexts, such as Spain, the EU and US in terms of maintaining and updating technical dossiers and CMC information, as well as reviewing promotional materials for compliant marketing.
Our company stays up-to-date in regards of the pharmacovigilance requirements and regulations. Also, considering the amount of information disseminated on social networks, we can help you monitor drug safety information that appears on social media sites with a new automatic Active Listening System.
Related Resources

The Electronic Application Form (eAF) is a unified EU form used for human and veterinary MAAs, variations, and renewals. It combines PDF for input and XML for data transfer, enabling data import/export, auto-copy, self-validation, and access to standardized term catalogues (EUTCT)—streamlining regulatory submissions and reducing manual input.

Highlights video

Highlights video

Highlights video

Highlights video

Highlights video

Highlights video

A case study about Marketing Authorization Expansion in Europe. A small European
Pharmaceutical company with
local activity in its country, with a
growth strategy aiming to
expand to international
markets but lacking experience
and capability in managing
complex EU procedures.
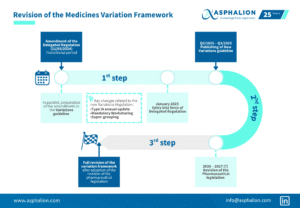
Embrace the Power of Smart Regulatory Strategy during this week! At Asphalion, we are dedicating this week to delving into the field of Life-Cycle Management.

Embrace the Power of Smart Regulatory Strategy during this week! At Asphalion, we are dedicating this week to delving into the field of Life-Cycle Management.

Embrace the Power of Smart Regulatory Strategy during this week! At Asphalion, we are dedicating this week to delving into the field of Life-Cycle Management, starting today with this infographic about the electronic application form (eAF).
Services
SDGs
As a company committed to sustainability, we follow the UN’s 2030 Agenda and its Sustainable Development Goals.
Memberships & PartnershipS









Quality Management System


Innovative SME

SDGs
As a company committed to sustainability, we follow the UN’s 2030 Agenda and its Sustainable Development Goals.
© Asphalion
Schedule here a free 30-minutes meeting with one of our consultants and tell us about your project, challenges or doubts.
We will be happy to assist you!

| Cookie | Duration | Description |
|---|---|---|
| cookielawinfo-checkbox-advertisement | 1 year | Set by the GDPR Cookie Consent plugin, this cookie records the user consent for the cookies in the "Advertisement" category. |
| cookielawinfo-checkbox-analytics | 1 year | Set by the GDPR Cookie Consent plugin, this cookie records the user consent for the cookies in the "Analytics" category. |
| cookielawinfo-checkbox-necessary | 1 year | Set by the GDPR Cookie Consent plugin, this cookie records the user consent for the cookies in the "Necessary" category. |
| cookielawinfo-checkbox-others | 1 year | Set by the GDPR Cookie Consent plugin, this cookie stores user consent for cookies in the category "Others". |
| CookieLawInfoConsent | 1 year | CookieYes sets this cookie to record the default button state of the corresponding category and the status of CCPA. It works only in coordination with the primary cookie. |
| elementor | never | The website's WordPress theme uses this cookie. It allows the website owner to implement or change the website's content in real-time. |
| viewed_cookie_policy | 1 year | The GDPR Cookie Consent plugin sets the cookie to store whether or not the user has consented to use cookies. It does not store any personal data. |
| wpEmojiSettingsSupports | session | WordPress sets this cookie when a user interacts with emojis on a WordPress site. It helps determine if the user's browser can display emojis properly. |
| Cookie | Duration | Description |
|---|---|---|
| _ga | 1 year 1 month 4 days | Google Analytics sets this cookie to calculate visitor, session and campaign data and track site usage for the site's analytics report. The cookie stores information anonymously and assigns a randomly generated number to recognise unique visitors. |
| _ga_* | 1 year 1 month 4 days | Google Analytics sets this cookie to store and count page views. |
| _gat_gtag_UA_* | 1 minute | Google Analytics sets this cookie to store a unique user ID. |
| _gid | 1 day | Google Analytics sets this cookie to store information on how visitors use a website while also creating an analytics report of the website's performance. Some of the collected data includes the number of visitors, their source, and the pages they visit anonymously. |
| Cookie | Duration | Description |
|---|---|---|
| VISITOR_INFO1_LIVE | 6 months | YouTube sets this cookie to measure bandwidth, determining whether the user gets the new or old player interface. |
| VISITOR_PRIVACY_METADATA | 6 months | YouTube sets this cookie to store the user's cookie consent state for the current domain. |
| YSC | session | Youtube sets this cookie to track the views of embedded videos on Youtube pages. |
| yt-remote-connected-devices | never | YouTube sets this cookie to store the user's video preferences using embedded YouTube videos. |
| yt-remote-device-id | never | YouTube sets this cookie to store the user's video preferences using embedded YouTube videos. |
| yt.innertube::nextId | never | YouTube sets this cookie to register a unique ID to store data on what videos from YouTube the user has seen. |
| yt.innertube::requests | never | YouTube sets this cookie to register a unique ID to store data on what videos from YouTube the user has seen. |
